A cascade of genetic information is drowning science. Sequencing machines decode plant and animal DNA so quickly computers can’t analyze the resulting data fast enough. It’s especially true for genome assembly, the final step in compiling the strings of letters that represent every creature’s genetic instructions.
Now researchers from Lawrence Berkeley National Laboratory and the University of California, Berkeley, have developed an assembly program so efficient they believe it could handle output from all the world’s sequencers on just part of one supercomputer.
The team will report on its new program in a paper presentation Nov. 17 at the SC15 supercomputing conference in Austin, Texas.
The efficient code will help geneticists advance science in a range of areas, from finding bacterial genes that produce drugs, biofuels and more, to identifying mutations behind cancer and other diseases.
Gene sequencing analyzes an organism’s DNA, comprised of four chemical bases represented by the letters A, C, G and T. The letters are instructions for cells to produce proteins, life’s workhorse molecules.
To rapidly sequence the billions of bases in a single organism’s genes, scientists break copies of a DNA strand into short pieces, or reads. Machines distribute the reads to multiple sequencers that decode them into the A, C, G and T bases. Today’s sequencers can decode a genome in hours, rather than the months or years it took in genetic research’s early days.
The whole process is like shredding a huge book, then reassembling it in correct order. But the reassembly step is a bottleneck in the process. To put the reads back in the correct, accurate order, high-performance computing (HPC) systems match overlaps in their letter patterns. It can take from a day to a week, depending on the computer’s speed and the genome’s size.
Researchers at the DOE Joint Genome Institute (JGI) at Berkeley Lab developed their own assembly pipeline program, called Meraculous (taking its name from k-mer, a DNA sequence containing a set number, k, of bases). Meraculous produces high-quality results but can take up to two days to assemble a 3-billion-base human genome, says Evangelos Georganas, a Berkeley Lab researcher and UC Berkeley graduate student in electrical engineering and computer science. Assembling bigger genomes, like those of the wheat plant or pine tree, can take as much as a week.
But by rewriting Meraculous, Evangelos and a team from JGI, UC Berkeley and Berkeley Lab can now assemble a human genome in as little as 8.4 minutes. And that’s using just part of Edison, a Cray XC30 supercomputer at the lab’s National Energy Research Scientific Computing Center (NERSC).
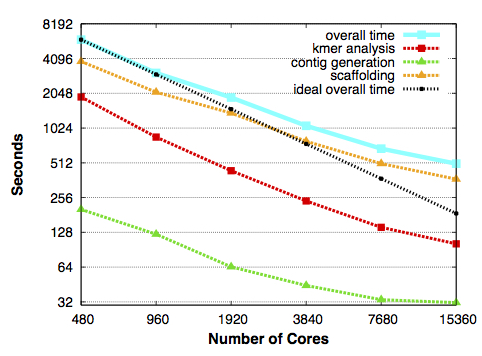
Scaling for HipMer and its components, with the run time in seconds falling in direct proportion to the number of processing cores it used on Edison, NERSC’s Cray XC30 supercomputer. The black dashed line shows overall scaling under ideal conditions. From Evangelos Georganas, Aydın Buluç, Jarrod Chapman, Steven Hofmeyr, Chaitanya Aluru, Rob Egan, Leonid Oliker, Daniel Rokhsar and Katherine Yelick, “HipMer: An Extreme-Scale De Novo Genome Assembler”. 27th ACM/IEEE International Conference on High Performance Computing, Networking, Storage and Analysis (SC 2015), Austin, TX, USA, November 2015.
The program also is scalable: As it runs on more computer processors, it works faster and can handle bigger genomes in the same amount of time.
But assembling the human genome in under 10 minutes isn’t what’s important, Georganas says. “Being able to provide new ways to do science” is the real achievement. Assembling multiple genomes in an hour and experimenting with varying parameters lets geneticists easily test different assumptions. “That, at the end of the day, would make them better understand the results they get.”
The new program, called HipMer (high-performance Meraculous), and Meraculous are de novo assemblers: They start from scratch, without a previously sequenced genome to refer to for guidance. De novo assembly is like constructing a jigsaw puzzle without seeing the picture on the box.
Meraculous first cuts the DNA reads into k-mers, each a fixed length of k bases. Its algorithm removes erroneous genome reads and puts the k-mers in a graph representing their overlapping sequences. The program iteratively searches the graph to find overlapping k-mers and build them into longer pieces called contigs. The program then uses data from the original DNA reads to order and orient the contigs, then stitch them into longer segments called scaffolds. Finally, Meraculous taps the read data to close gaps between scaffolds and create a full genome.
In supercomputers, much of this work typically is done via parallel processing: Programs divide tasks and distribute the parts to individual processor cores. With cores tackling each part simultaneously – in parallel – the computer reaches an answer more quickly than performing tasks one at a time (in serial).
Parts of Meraculous were parallel but inefficient, Georganas says. Meanwhile, the step to analyze the graph and assemble k-mers into longer contigs and scaffolds was programmed serially and consumed lots of working memory. He and his colleagues, including Berkeley Lab’s Aydın Buluç, Steven Hofmeyr and Leonid Oliker, set out to fully parallelize the code. Other collaborators include JGI’s Jarrod Chapman, Rob Egan and Daniel Rokhsar and UC-Berkeley’s Chaitanya Aluru.
In one of the first steps, the team converted much of Meraculous from the Perl language to Unified Parallel C (UPC), a Berkeley Lab-designed extension of the common C language. “Just writing the code in C gives you some significant speed-up,” Georganas says. “And when you parallelize the C code (as in UPC), it goes even faster.”
UPC also treats the computer’s memory as a single, global address space. “You don’t have to bother with which processor owns what data,” as in some other approaches, Georganas says. That’s especially useful when communication between processors and memory is irregular – as when the program is finding overlapping k-mers. Georganas’ academic advisor, Katherine Yelick – Berkeley Lab’s associate director for computing sciences and recipient of an SC15 award for helping set the course in computing research – is one of UPC’s creators and a co-author on the SC15 paper.
The initial assembly applies probabilistic filters to remove erroneous k-mers, a process that requires counting each occurrence of each k-mer. There can be only one of one k-mer and millions of another, so the work often is unevenly distributed between processors, slowing execution. HipMer breaks this barrier by identifying the highest-count k-mers and using multiple processors to count them.
To accelerate contig and scaffold assembly, HipMer uses hash tables, a way to index information so the system can find it efficiently. The algorithm uses a formula to assign a value to each piece of data, like a k-mer or a graph edge that connects k-mers. That value is stored in a hash table.
Metagenome sequencing can help scientists understand how microbes interact and which genes are most important for survival.
“We want to store them in a structured way such that when we ask for a particular vertex, we can find the hash value of that vertex and look it up in the table,” Georganas says. Because genomes are so enormous, the resulting hash tables also are huge – too big to be stored in the memory of a single supercomputer node (a group of processors that share memory).
“We can distribute this hash table to more computer nodes,” Georganas says. When the algorithm seeks a particular k-mer, it “might live in a part of a hash table in my node or it might live in a node that’s remote,” leading to irregular communication.
“This is where UPC is really convenient, because you don’t have to bother with where that k-mer lives. UPC gives you the notion of having all the k-mers in a single address space,” reducing the time the supercomputer spends trading information between nodes. “Once the data are stored and indexed in an efficient way, you can really speed things up,” Georganas says.
But that communication also can slow genome assembly. To address this, HipMer predicts how k-mers will be placed into contigs and partitions them so those belonging to the same contig are mapped to the same processor, reducing communication between nodes.
These and other changes to Meraculous have generated astonishing results. In tests described in the SC15 paper, HipMer assembled a human genome and a wheat genome on 15,360 Edison processing cores. The human genome took 8.4 minutes; the wheat genome, with around 17 billion bases, took 39 minutes.
The researchers reported similar results at last year’s supercomputing conference. Since then they completed parallelizing the entire code and making other changes.
The SC15 paper also includes preliminary results from a HipMer test on a huge task: assembling a metagenome, in which all the DNA from thousands of microbes found in an environment (such as soil or ocean water) is simultaneously sequenced and reassembled. Metagenome sequencing can help scientists understand how microbes interact and which genes are most important for survival. The results could advance biofuel development and other applications.
“When you go to the metagenome case, the data set, in size and complexity, explodes,” Georganas says. It becomes a question of not just whether it’s possible to assemble genomes quickly, but whether they can be assembled at all. “By running in parallel and using all the memory available in your system efficiently, you can pretty much break that barrier.”
The team assembled a metagenome sampled from an island in California’s Sacramento-San Joaquin River delta. In a limited run comparing performance on 10,000 cores and 20,000 cores, k-mer analysis and contig generation scaled, running faster on more cores, but data input and output did not.
It’s the next horizon for Georganas and his colleagues. “The bulk of the work is reusing the existing parallelized components from HipMer” and adding other already existing algorithms, he says. “At the end of the day, with minimal extra components, we can have a metagenome assembly pipeline fully parallelized.”
For now, Georganas and the team calculate that a single computer the size of Edison could handle with all the single-genome data from the world’s gene-sequencing machines. With their parallel methods, they’ve shown “de novo genome assembly is not a bottleneck anymore. We can keep up with the pace of getting more and more data – of course, assuming we have a supercomputer to run on.”
JGI researchers are using HipMer already and, after some testing, the team plans to release it as an open-source code. A URL where others can download it should be available soon.
The COVID-19 response demonstrated how computational biology could enhance public health research. Though the pandemic… Read More
Diamonds aren’t just beautiful sparkly rocks or a girl’s best friend; they’re also the hardest… Read More
More than a century ago, scientists pondered how evolution might be driven by mutations caused… Read More
Scientists, industries and policymakers have been working for years to find energy storage technologies that… Read More
The human brain contains a vast expanse of unmapped territory. An adult brain measures only… Read More
Computer scientists are democratizing artificial intelligence, devising a way to enable virtually anyone to train… Read More
{kind=link}