Electrifying transportation and storing renewable energy require improving batteries, including their electrolytes. These fluids help lithium ions shuttle seamlessly from anodes to cathodes. But if the electrolytes dry up or react with other components, batteries fail.
To design better battery electrolytes, Venkat Viswanathan of the University of Michigan and his team have developed and worked with a variety of computational tools. Now they are training a large language model (LLM) that could help researchers find recipes for these fluids and could be applied to many other chemistry problems from medicines to materials, with an allocation from the Department of Energy’s Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program.
As in sports drinks, electrolytes in batteries are mixtures of salts and liquids that help carry ions back and forth. Here they ferry lithium ions from the cathode to the anode as batteries are charged and in the reverse direction as they’re discharged.
In lithium-ion batteries, electrolyte fluids are organic solvents — primarily comprising carbon, hydrogen and oxygen atoms. As researchers optimize electrolytes, they include additives with important properties. They’re looking for a recipe that optimizes flow and ion movement but is also chemically inert and resists evaporation. So they’re seeking components with specific physical properties such as low viscosity and volatility and high stability.
When trying to develop new molecular mixtures, researchers often grapple with the mindboggling number of possible compounds that could exist (up to 1060). Theoretically, 50 billion molecules could be synthesized in a laboratory; perhaps 10,000 compounds have been studied for batteries, Viswanathan says. “It’s a huge gap between the number of possible options and how many have just been tested.”
Synthesizing and testing so many molecules is impossible, but computational tools offer ways to screen candidate molecules and model their properties. When computational researchers identify promising molecules in silico, experimental chemists can then focus their laboratory work on those likely candidates.
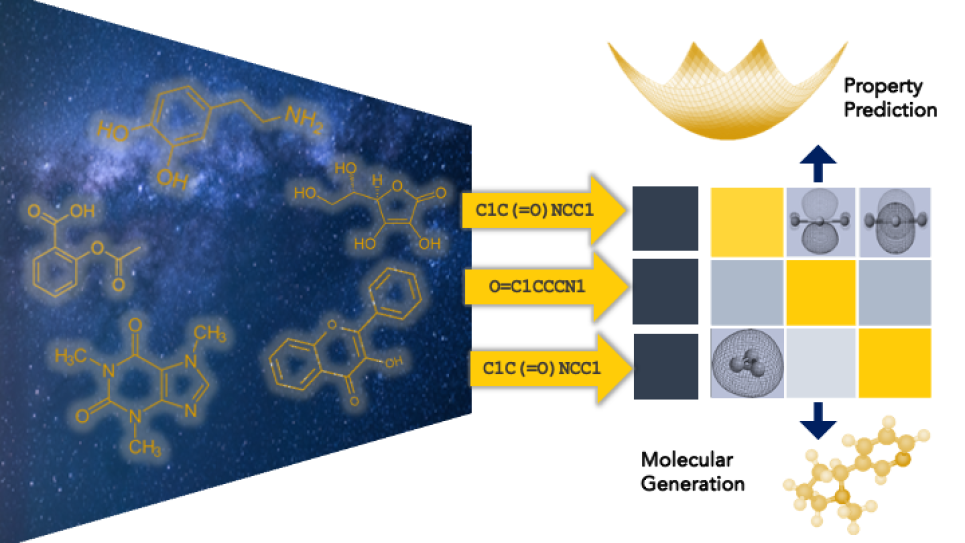
In recent years, machine learning methods have taken a central role in predicting molecular behavior. Traditionally those methods have been graph-based, Viswanathan notes, incorporating a molecule’s 3D structure into models that rely on image-classification. The models are trained on libraries of Tinkertoy-like chemical structures and their properties to predict new component combinations for electrolytes.
But with the success of ChatGPT and other LLMs, Viswanathan wondered if they could adopt conversation-assembling technology to build new compounds. LLMs use masked-language models, an approach that works like a fill-in-the-blank question: It blocks out part of a sentence and asks the model to fill in the gaps. Chemists have already developed tools, such as SMILES, that convert 3D structures of organic molecules into strings of letters that represent connected atoms. “The exact same machinery that is being used in natural language processing can be leveraged to do molecular discovery,” Viswanathan says.
‘We’re nowhere near the plateau. So we expect that once we train at that 50 billion scale, it would unlock new capabilities.’
But he also notes that the idea was somewhat counterintuitive because the text-based models omit 3D data that’s been considered essential for understanding how molecules fit together. Researchers have previously tried building chemistry-based LLMs but with smaller datasets. For example, ChemBERTa was trained on 60 million molecules and IBM’s MoLFormer on a billion compounds. While interesting, these earlier models did not outperform graph-based models.
Viswanathan suspected that a chemistry model built on an LLM-like foundation, or foundation model, would need more training data. “The core idea is that there are these so-called scaling laws, which is that when you have larger and larger training data sets, then you can now get models that are much, much more precise.” Based on molecular scaling laws, he and his team reasoned that a foundation model trained on 50 billion synthetically accessible compounds would perform well.
However, the team needed leadership-class computing resources to take on this gigantic training task, and that’s where the INCITE allocation came in. With 200,000 node-hours on Argonne Leadership Computing Facility’s Polaris, they could scale the model to incorporate all compounds from two existing chemical databases, REAL space and CHEMriya.
The project team includes collaborators contributing distinct expertise. Co-PI Arvind Ramanathan of Argonne National Laboratory successfully built an analogous LLM, GenSLM, for predicting DNA mutations. Bharath Ramsundar of Deep Forest Sciences led the earlier ChemBERTa model. To speed the training process, Natalia Vassilieva of Cerebras Systems assists with the use of advanced AI-accelerating hardware on Polaris.
So far Viswanathan and his colleagues have pre-trained their model on 2 billion molecules, twice the number of IBM’s MolFormer. “We’re seeing very, very strong progression,” he says. “We’re nowhere near the plateau. So we expect that once we train at that 50 billion scale, it would unlock new capabilities.”
This pre-training phase is an initial step that links molecules and fundamental chemical properties such as viscosity. Later they’ll fine-tune the model with electrolyte-specific datasets to work on the battery problem. In the future, researchers could optimize the initial model for other chemistry applications, such as drug discovery, designing fragrances or separating compound mixtures.
These computational results lay the foundation for experiments with robotic systems such as Clio, which Viswanathan’s collaborator, Jay Whitacre of Carnegie Mellon University, has developed to measure the physical properties of new mixtures and then test the promising ones within a battery.
Existing computational models are already accelerating the pace of research on new battery electrolytes. Viswanathan estimates that existing models have accelerated electrolyte research by two- to five-fold, and he anticipates that a comprehensive foundation model could further increase research productivity by a similar amount.
Besides speeding the search for promising electrolytes, foundation models may go even further. For example, a successful foundation model for chemistry could be connected to a more general LLM. This summer, Viswanathan received a National Artificial Intelligence Research Resource Pilot Award from the National Science Foundation to pursue this idea. He envisions a merged model that experts and non-experts alike could enlist to probe chemistry questions with text prompts like those used with ChatGPT.
So far, AI-based models have not reached the threshold of making a scientific discovery that impresses humans with its novelty, but foundation models could help close that gap. He says, “The promise of these foundation models is it’ll actually get you to an answer you would have never gotten to.”