Over the past two years, artificial intelligence has shown it can predict what many cellular components look like. For instance, the AlphaFold deep-learning tool developed by Google sister company DeepMind has decoded how nearly every amino acid sequence folds into defined shapes.
With a grant of computing time from the ASCR Leadership Computing Challenge program, a team led by Jeffrey Skolnick of the Georgia Institute of Technology, is extending that work to unfurl how those proteins interact and form complex, working structures in living systems.
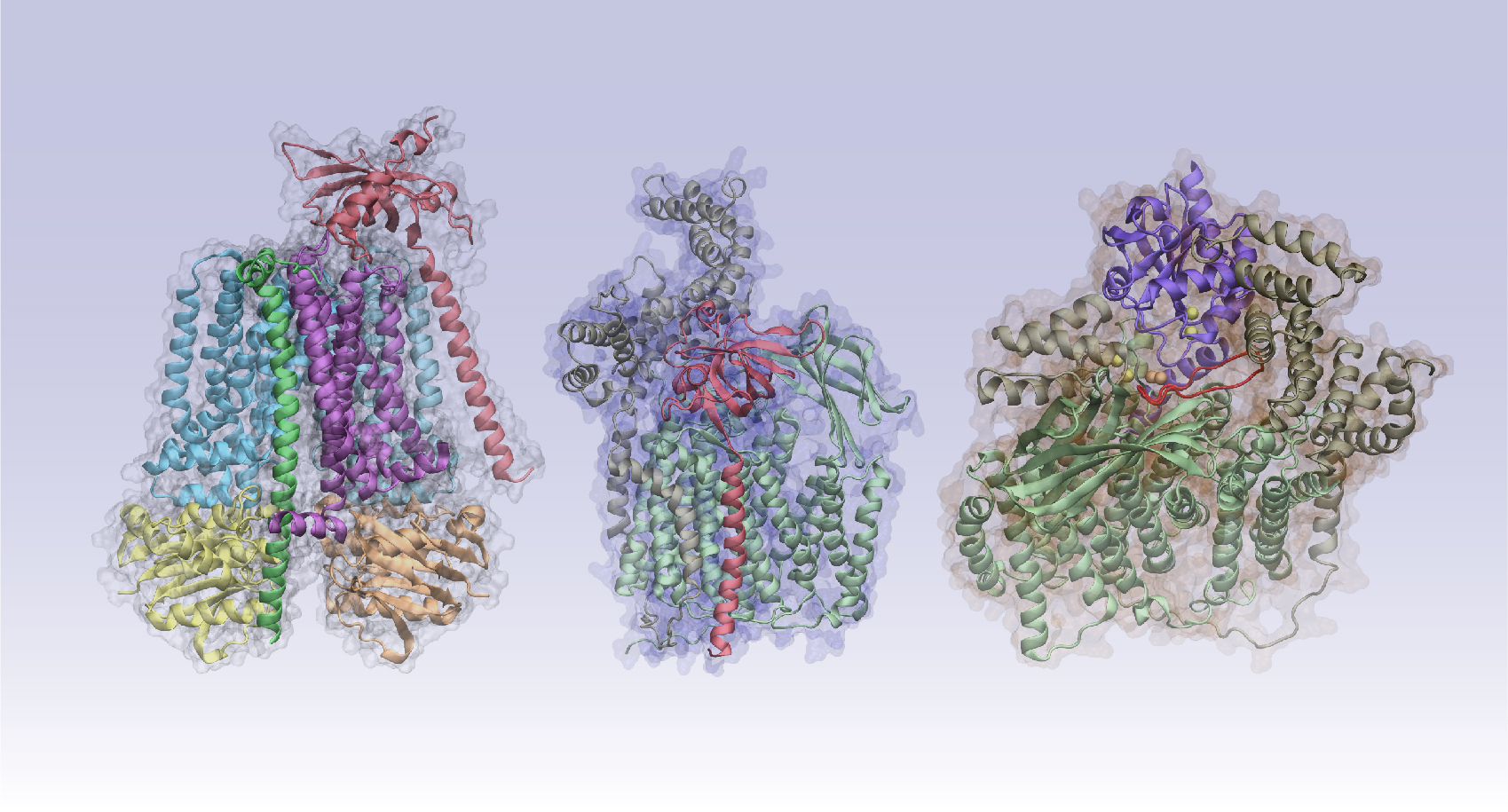
Ascertaining a protein’s structure is the first step in determining its function. Most are pieces of complex arrangements and observing an isolated structure won’t show “the difference between living systems and dead systems,” Skolnick says. In living systems, “the molecules interact.”
How the molecular parts mesh remains a biology grand challenge. Addressing it requires identifying which proteins interact and what the resulting structures resemble. That second challenge can bring in other biological and functional concerns, he says, such as whether binding between two partners interferes with binding to others.
Skolnick, Georgia Tech scientist Mu Gao, Oak Ridge National Laboratory (ORNL) scientist Jerry Parks and their colleagues started with the now open-source AlphaFold algorithm, modifying it to better tackle complex structures.
The team incorporated a tool that rates how well the surfaces will stick together.
The original algorithm uses pairwise alignments, in which sequences are added and aligned stepwise to build a pattern. That feature “implicitly tells you something about the structure,” Skolnick says. But it doesn’t tell him “how they interact. And, moreover, I would like to be able to feed in six of these sequences, nine of those sequences and 35 of the other ones, put it all together in the soup and have them assemble into the complex.”
The team’s modified AlphaFold, called AF2Complex, has a built-in unpaired-alignment strategy that is twice as accurate as traditional docking-based approaches for predicting protein-protein interactions. The team also incorporated an interfacial similarity score – a tool that rates how well the surfaces will stick together.
“They can be structurally complementary, but if the residues don’t like being in the interface, they’re not going to interact,” Skolnick says. For example, two negatively charged surfaces will repel each other even if their shapes match. The score rarely produces false positives, and it allows researchers to quantify how well they trust the prediction – whether the interactions highlighted are most likely random, potentially interesting or nearly certain. “None of these algorithms works a hundred percent of the time. What you want are complementary approaches. As long as you know when they are right and when they are wrong, that’s the key.”
Other team members include Georgia Tech undergraduate Davi Nakajima An, ORNL’s Ada Sedova and Mark Coletti and the University of Missouri’s Jialin Cheng. Even with allotments on two of the world’s most powerful computers, ORNL’s Summit and the National Energy Research Scientific Computing Center’s Perlmutter, such simulations are limited to modeling systems of up to 5,000 amino acids because their innumerable individual atoms can push the limits of graphics processing unit memory.
To predict how these proteins function, the team modeled protein-complex assemblies and their role in cell-signaling pathways. In an April Nature Communications paper, the researchers examined the protein complexes in the cytochrome C pathway. The protein is found in mitochondria, organelles critical to energy production in cells. The authors not only predicted structures that had been discovered experimentally but also generated a previously unknown structure. Their predictions included placing the iron-binding heme group in roughly the correct location. That’s a notable improvement over publicly available AlphaFold versions, Skolnick says.
More recently they have explored how Gram-negative bacteria’s outer membrane proteins (OMPs) travel from inside a cell – where they’re produced – to its surface. “Somehow it crosses two membranes, a periplasmic region [between the two], and it gets stuck on the outer membrane.”
The team observed that one of these chaperone proteins and the OMP only make minimal contact with each other. “It’s a plausible model,” Skolnick says, because it costs energy to build and break short-lived interactions between these proteins. This minimalist strategy could make sense as cells shuttle the proteins to their surfaces.
Experimentalists must test this and other predictions to determine their accuracy, Skolnick says. “All the work we’re doing is trying to create a prioritized list for experimentalists of ‘look here, not there.’ If you’re interested in a particular biochemical pathway or function, why not start with the more confident ones and work your way down?”