For a common virus, influenza remains an enigma. Some strains – like the one that caused the 1918 pandemic – can kill, whereas others cause an inconvenient fever. And perhaps more curious yet is how flu mutates, forcing drug developers to come out with new vaccines year to year that may or may not work as hoped. But what if they could boost a vaccine’s reliability by targeting more stable parts of the virus?
Researchers are putting such questions to supercomputers, studying influenza to not only understand how the virus works but also to devise therapies that circumvent the mutation conundrum.
Biophysicist Rommie Amaro of the University of California, San Diego (UCSD), has been studying flu and how computational models could aid drug discovery for the past decade and a half, first as a postdoctoral fellow working with Andrew McCammon, a UCSD theoretical chemist.
Around the time Amaro began, in 2005, scientists had just resolved partial structures of the flu virus using a technique called X-ray crystallography. It revealed the relative position of atoms in the bug, allowing researchers to numerically encode each and apply equations to describe the interactions between different atoms in the system.
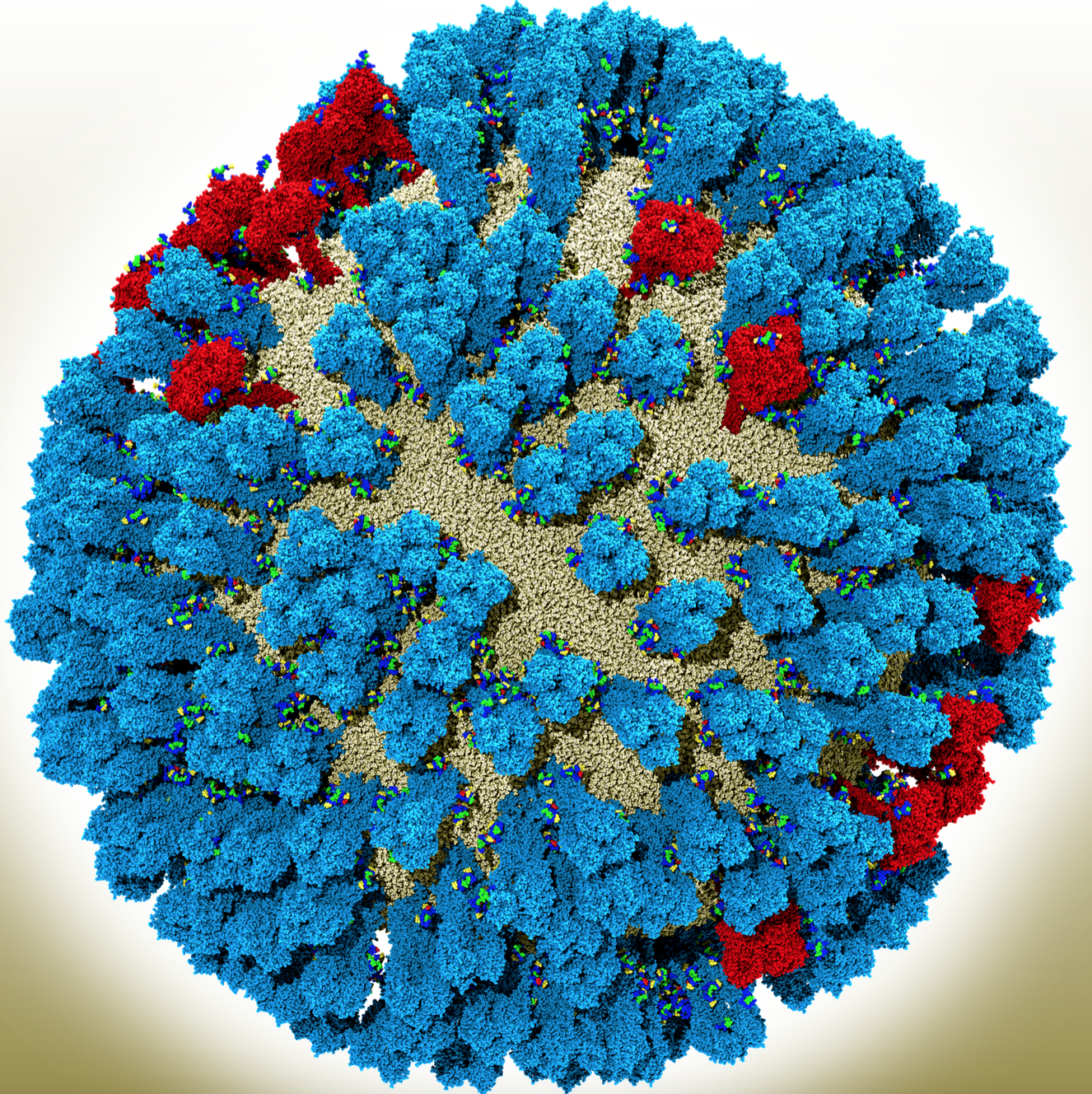
During her postdoc, Amaro studied a viral-surface-coating protein called neuraminidase (NA) that allows influenza to replicate inside cells. NA and three other identical units assemble into one structure. Before researchers could scrutinize influenza on supercomputers, they had snapshots of the NA proteins frozen in time. What they didn’t know was how they transitioned from one state to another.
Over the past decade or so, technological advancements have given flu researchers a way to study the dynamics of a single protein, or a single state, to model the virus’ true biological complexity. Amaro, for instance, has used a supercomputing allocation from the Department of Energy’s INCITE (Innovative and Novel Computational Impact on Theory and Experiment) program. “Protein dynamics matter in other systems, like cancer,” Amaro says. “We’re trying to (show) the same for flu.” She and her colleagues are studying the H1N1 influenza virus –responsible for the 2009 swine-bird flu pandemic – on Titan, the Cray XK7 supercomputer at the Oak Ridge Leadership Computing Facility, a DOE national user facility.
‘This could allow us to develop more durable therapies for flu.’
Researchers use a technique called Markov state model theory to analyze data gathered from these models. The approach describes the dynamics of individual proteins and then combines these paths into a single analysis. This allows researchers to extract long-timescale dynamics from many short-timescale simulations.
For influenza, this means being able to visualize and simulate the entire viral-membrane coat and to see how different proteins interact. Besides NA, the flu virus surface is coated with another protein called hemagglutinin (HA). In these full-virus simulations, researchers can see how both the HA and NA – which belong to a class called glycoproteins – jostle each other, affecting the dynamics of the virus, Amaro says.
She and her colleagues have simulated the whole virus, with its covered viral coat. “With so many [NAs and HAs] in one system, we can now rigorously characterize the kinetics of the motion that we see,” she says. By studying the protein kinetics, Amaro and her colleagues have found new pockets that drugs could target. “This could allow us to develop more durable therapies for flu,” she says.
The position and dynamics of glycoproteins also matter when drug developers think about making a universal vaccine. Though influenza vaccines typically target the head of the HA molecules, computational models show the virus’ universal antigen is actually on HA’s side. Only by observing the structure of the full influenza virus, Amaro says, can researchers and drug developers understand how difficult it would be for an antibody to access that site. “We’re starting to get the first views into that with these larger-scale models.”
Another puzzling aspect of flu: As the virus circulates in people, sugars called glycans collect on its surface, often latching onto HA units. Influenza vaccines, meanwhile, also target the HA portion of the virus. When the virus adopts new glycans every six years or so, that invariably affects the dynamics of the virus, sending drug developers back to the drawing board for a new vaccine.
Amaro and her colleagues are using Titan to investigate how the H1N1 virus behaves with glycan attachments. “It’s the first time we can see what’s happening with the glycans at the atomic level, since they vary in complexity,” Amaro says. “These simulations will give a sneak peek into what these glycans might look like and how they affect the dynamics of proteins of the virus.” Understanding how glycans alter the virus’ dynamics or the structure of the active site could inform design of a drug that could bind well even with a few glycans present.
Amaro is also modeling how the H1N1 virus with glycans binds to its native substrate, sialic acid, which is found on host cells. Before the virus is able to enter, HA must bind to sialic acid and NA must cleave it. By looking at the virus with all its glycan attachments, Amaro and her colleagues will able to get a sense of protein dynamics once the native substrate is introduced. Pharmaceutical companies also have developed NA-binding influenza inhibitors intended to stop the virus from replicating in the body. (Vaccines are useless once someone’s infected.) Amaro hopes that the kinetics of these structures can guide others who devise new influenza inhibitors.
For instance, Amaro collaborates with structural biologist Ian Wilson and chemist Dennis Wolan of Scripps Research in La Jolla, California. They’re basing drug-design experiments on Amaro’s structural dynamics results to test the simulations’ therapeutic relevance.
Amaro hopes to build a model host-cell and use similar molecular dynamic techniques to visualize how the influenza virus interacts with it. “This would enable us to understand the infection process in a way we would never be able to characterize experimentally. If we had a drug, we could see how it changes the infection process.”