For years, computational biologists have sought ways to model how proteins change shape in real time. In the past decade, they’ve glimpsed the possibilities as new programs and powerful computers came on line, letting researchers learn more about disease mechanisms that once eluded them.
That includes novel techniques to simulate dynamics of proteins involved in autoimmune diseases, the interest of Rice University’s Cecilia Clementi, a computational biophysicist, chemistry professor and senior scientist at Rice’s Center for Theoretical Biological Physics.
With groups headed by Frank Noé at Freie Universität in Berlin, Shantenu Jha at Rutgers University in New Jersey, and Jeremy Smith at the Department of Energy’s Oak Ridge National Laboratory in Tennessee, Clementi’s Houston-based team has focused on the major histocompatibility complex class II (MHC II), which helps our immune system distinguish disease-related protein fragments from those belonging to our own cells.
Before our immune system can attack a pathogen, such as a bacteria or virus, our bodies first process these intruders into smaller protein parts called antigens inside specialized antigen-presenting cells. Once the antigen is properly bound to the MHC-II protein on the cell surface, it signals to T-cells, which ultimately fight the infection.

Cecilia Clementi. Photo courtesy of Jeff Fitlow/Rice University.
In autoimmune conditions, however, the body’s immune response has difficulty seeing the difference between foreign cells and self. Researchers have long suspected an error in loading the proper antigen onto MHC II is behind most autoimmune conditions, such as type 1 diabetes, multiple sclerosis, rheumatoid arthritis and Crohn’s disease. But exactly what changes occur, and how, during this exchange remain unknown.
Autoimmunity therapies – including over-the-counter anti-inflammatory drugs, steroids, and newer biological antibodies – seldom target a condition’s root causes and come with wide-ranging side effects. Clementi and her colleagues hope that studying MHC-II protein dynamics, especially how the antigen is loaded onto the protein in normal and disease states, will ultimately help find new drug targets.
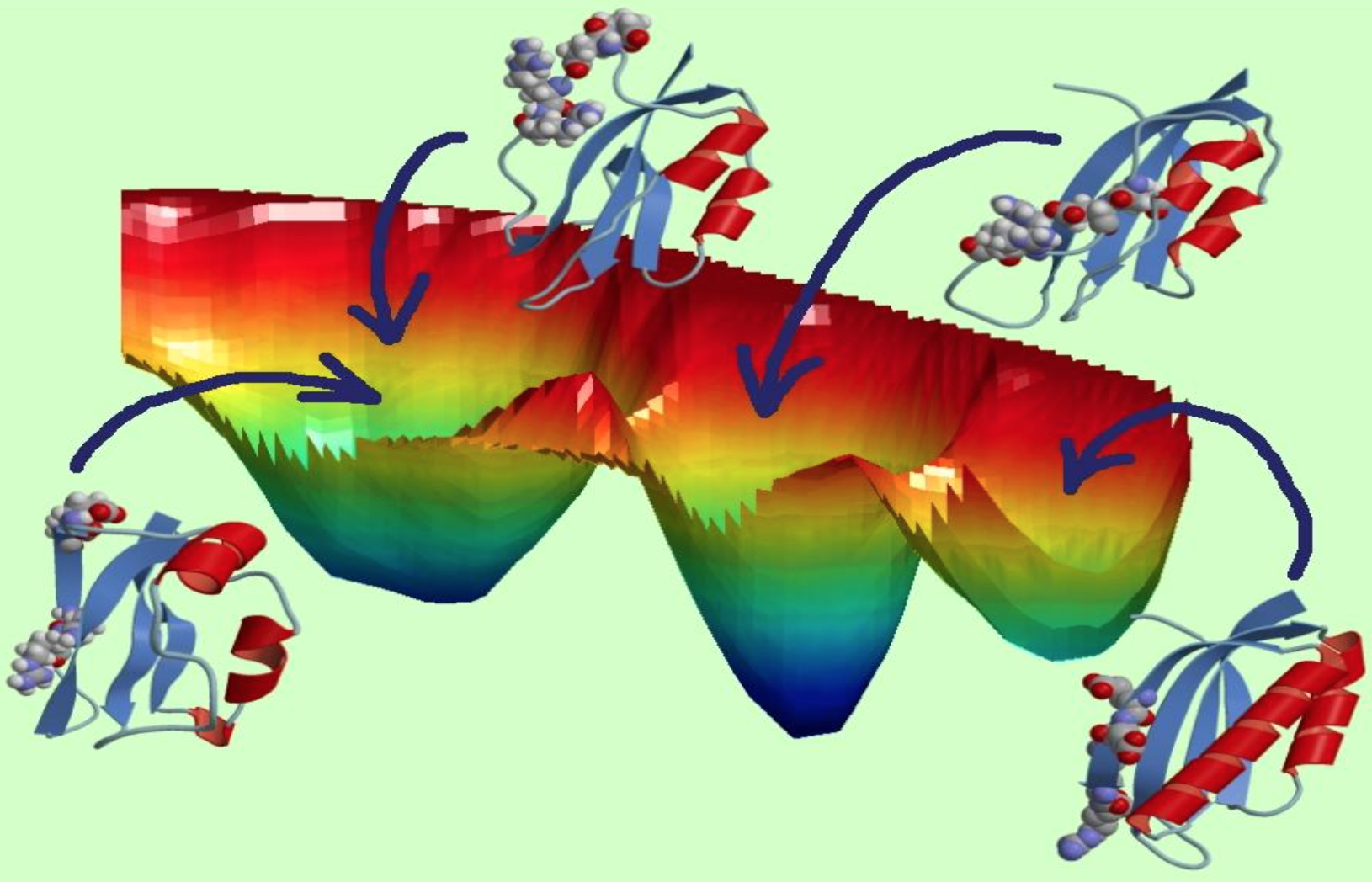
But the MHC-II protein is complex and all of its possible configurations are difficult to predict without extensive computational resources. The DOE’s INCITE program (for Innovative and Novel Computational Impact on Theory and Experiment) has supported Clementi and her collaborators since 2017 with millions of processor hours on DOE machines. So far, Clementi and her collaborators have logged 81 million processor hours on Titan.
Their approach, called Adaptive MD, generates a system that can iterate through a large number of possible protein conformations on thousands of graphics processing units – or GPUs, computer chips related to those used in video games. The method combines adaptive sampling, which periodically restarts many parallel simulations to explore a large number of protein structure configurations, and Markov state modeling, which enables analysis of shapes already sampled to determine where new simulations should be restarted. The team runs its algorithms on Titan, a Cray XK7 at the Oak Ridge Leadership Computing Facility, a DOE Office of Science user facility. Titan incorporates nearly 19,000 GPUs, letting computational biologists observe protein dynamics for microseconds – a timescale in which these biological processes unfold in nature.
‘These are massive simulations that we cannot do if not on Titan.’
Clementi likens the technique to scouting for a way across a mountain range without trails. “If you’re alone and don’t have a map, you will start walking in random directions, and it’s going to take forever. There are just too many paths. But if you have a large group of people who go off in all different directions but can still communicate with one another, they can tell each other what they’ve seen, which will dictate where they go the next day based on the most promising path.”
To date, Clementi and her associates have investigated the dynamics of 14 MHC-II variants. Noé’s group at Freie Universität found two mutants, beta N82A and alpha Q9A-alpha E11Q; experimental collaborators showed the mutants have biomedical significance. This work resulted in a 2016 paper in Nature Communications. Researchers had already found that beta N82A formed an unstable complex with HLA-DM, the protein that loads an antigen into MHC II.
Although crystal structures can be useful to better understand proteins, it’s becoming increasingly clear their overall dynamics – not just a single snapshot – are important to their function, Clementi says, and capturing information between the snapshot frames requires the most powerful high-performance computing. “These are massive simulations that we cannot do if not on Titan.”
Designing MHC-II–centered therapies is a long way from the basic science, but Clementi and colleagues’ early findings already suggest potential ways to target the protein.
“It’s clear that it’s not just one conformation that’s important,” she says. Instead, “it’s the tendency of the protein to explore different configurations that is modified in mutants. Since we know that dynamics are important, one could potentially think about using allosteric drugs –drugs that modify conformational switches – to target MHC in the future.”
Next, Clementi and collaborators want to use their computational methods to assess the dynamics of 21 other clinically relevant MHC-II mutants. They will also release their Adaptive MD software later this year.