From batteries to biology, how atoms move is critical for understanding how matter behaves. Unless temperature is lowered to absolute zero, atoms routinely bump into each other or fly apart. These critical interactions determine a material’s properties, such as how well it can store electricity or the energy of chemical reactions.
But accurate simulations of these systems’ detailed physics and chemistry requires astronomical amounts of computational power and time. Until recently, because of this complexity, researchers could perform detailed simulations of only simplified problems with just a thousand atoms.
Now, innovations from Department of Energy (DOE) national laboratory researchers are producing algorithms for these detailed calculations, known as first-principles molecular dynamics (FPMD). These new FPMD techniques streamline calculations to accelerate run time and boost efficiency, allowing researchers to simulate a million or more atoms. Last fall, their work was recognized as a finalist for the Gordon Bell Prize, the highest honor in high-performance-computing research for scientific applications.
Because atoms are smaller than a nanometer, researchers typically can’t directly observe what happens via experiments. Instead, they use computational models to understand molecular behavior in action. But it’s challenging because of the many variables involved in reproducing the complex behavior of multiple atoms interacting.
Historically, computational chemists have used a technique called classical molecular dynamics to study systems with thousands or millions of atoms, says Jean-Luc Fattebert, a computational physicist who led this work at the Lawrence Livermore National Laboratory (LLNL) Center for Applied Scientific Computing. He recently joined the Computational Sciences and Engineering Division at the DOE’s Oak Ridge National Laboratory. Classical molecular dynamics models approximate atom behavior as individual balls and use simple rules – for instance, the attraction of opposite charges and the repulsion of similar charges – to describe their interactions. Such simulations can be useful in many cases, he notes, “but these models are often not good enough to represent what’s going on.”
That’s because such simulations ignore the behavior of electrons, which are difficult to represent. Yet that information often is essential for accurately calculating what’s happening within groups of atoms.
The complexity in simulating electrons comes from quantum mechanics. Electrons aren’t classical particles that are easy to describe in space, Fattebert says. Instead, researchers must describe them as clouds of probability. Doing that requires a whole equation, not just a single point. Scientists also must consider how the electrons interact.
Those essential adjustments dramatically increase the computational complexity (the number of operations needed to solve for each equation) and as the number of atoms grows, the size of the calculation balloons like the cube of the number of atoms. For a single snapshot of these molecules, all the atoms also must be calculated for each step forward in time. To really understand what’s happening as molecules move and chemical reactions occur, researchers must assemble thousands of these time steps in succession to follow the atoms for tens of picoseconds (trillionths of a second).
If communications are not managed efficiently, computations slow to a crawl.
To accomplish this feat in a reasonable time – a few weeks or months on a supercomputer – these complex calculations must happen quickly, in a minute or less for each time step. Fattebert and his team are approaching that goal: They’ve successfully simulated more than one million atoms at 1.5 minutes per time step.
To boost the calculations’ speed and efficiency, the LLNL team built algorithms that helped reduce the computational intensity. Some details of how electrons interact – particularly when those electrons are far from each other – don’t change how the overall system behaves. So the team found ways to eliminate those extra terms. What’s more, their algorithm uses modern linear algebra solvers, mathematical tools that speed the calculations.
The new algorithms also streamline and prioritize communication between a supercomputer’s individual processing cores. These innovations were critical to take advantage of the most powerful supercomputers, such as Sequoia, a petascale IBM Blue Gene/Q with more than a million cores that LLNL acquired in 2012.
Just as electrons’ arrangement in space greatly influences chemical behavior, the calculations of individual electrons are distributed among a supercomputer’s individual cores. Communication between these parallel cores is critical for solving this type of computational problem, Fattebert adds. If it’s not managed efficiently, computations slow to a crawl.
In these simulations, interactions between electrons represent most of this communication. One trick to boost computational efficiency is to limit the amount of communication and the distance messages must travel. The team’s algorithms eliminate unnecessary communications between distant electrons and also trim messaging between processors. In addition, cores that are computing electrons located close to each other in space also are near each other on the supercomputer.
The work has garnered accolades, and the LLNL team has been working with researchers to implement these tools for a variety of research questions while still improving the algorithm. They work closely with scientists studying applications to ensure that various parameter adjustments produce accurate results. They’d also like it to become code that researchers can use routinely, Fattebert says.
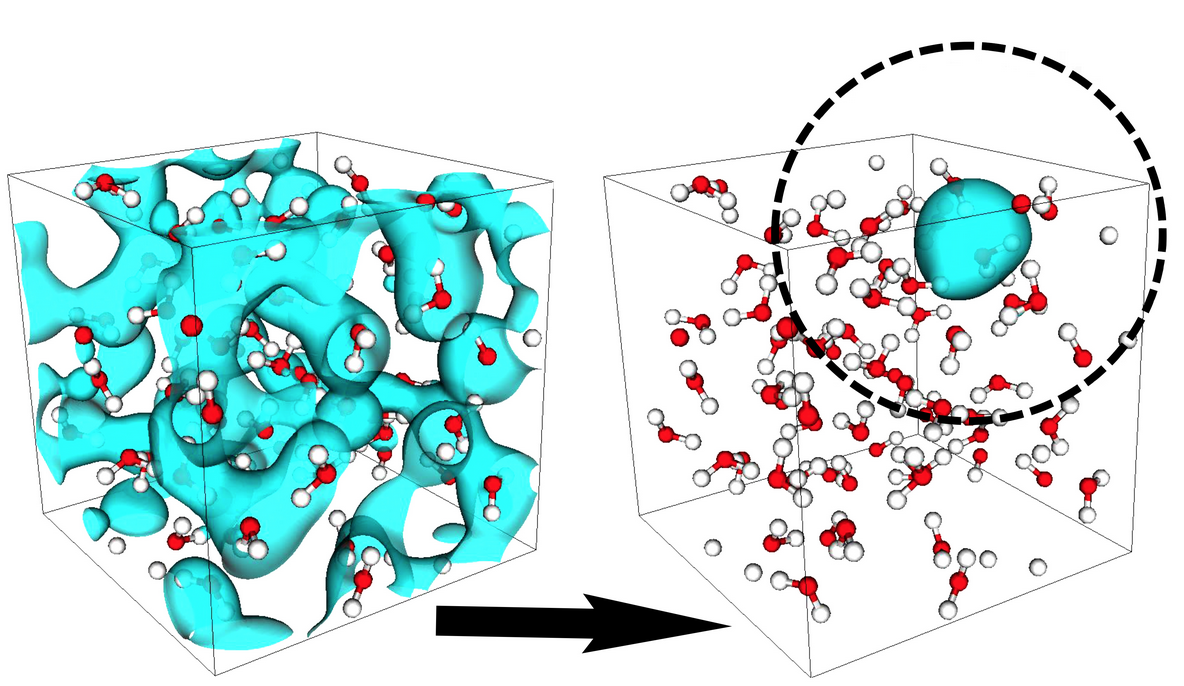
Besides making the algorithms more robust and easier for researchers to use, Fattebert and his team are extending their work to study a wider range of materials. So far, they’ve focused on insulators (materials that do not conduct electricity), particularly water because of its ubiquity and importance to a range of natural and synthetic systems. Molecules dissolved in water are everywhere, from energy storage to chemical reactions in biology.
Metals, which conduct electricity, represent a greater challenge, Fattebert says. In these materials, electrons interact with many more of their neighbors, greatly enhancing the computational complexity. “It’s harder to neglect these interactions between pairs that are far from each other.” Plus, their energetic behavior is far more complex to describe. The team is trying to tease apart these challenges to see if they can get their algorithms to work with such materials in the minute-per-step range.
Lawrence Livermore National Laboratory is managed by Lawrence Livermore National Security, LLC for the U.S. Department of Energy’s National Nuclear Security Administration.
Oak Ridge National Laboratory is supported by the Department of Energy’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.