Computational chemists have made strides modeling many complex chemical interactions, thanks in part to massively parallel computing and algorithms that approach predictive accuracy. But even the best methods trade off between atomic-scale accuracy and simulating how thousands of atoms behave. No chemistry simulation has accounted for the attraction and repulsion of each molecular orbital because the job has been too computationally demanding.
Bridging this gap has been high on the to-do list for computational chemists worldwide, including a collaborative group from Denmark and Oak Ridge National Laboratory, which reports nearing that goal. Computational chemists, led by Poul Jorgensen of Aarhus University, are using the Titan supercomputer at the Department of Energy’s Oak Ridge Leadership Computing Facility to break computing records for modeling a molecular nanowire system containing thousands of atoms.
The research, with support from a DOE Office of Science Innovative and Novel Computational Impact on Theory and Experiment (INCITE) award, involves a class of molecules called supramolecular wires – nanoscale filaments that self-assemble in solution from subunits and are cemented by local interactions among electrons in the subunits.
These intermolecular forces arise when individual electrons temporarily become unbalanced in an atom and form a fleeting positive or negative charge that can attract or repel a nearby atom. Known as van der Waals forces, they’re often described in chemistry textbooks as the weakest molecular forces.
But don’t tell that to the gecko, which uses those very forces to cling to and scale sheer vertical surfaces. If molecules are densely packed, as they are on the millions of tiny hair follicles lining the gecko’s footpad, these interactions can become coordinated and quite strong.
Indeed, among the chemical interactions that occur in a supramolecular wire assembly, weak forces are critical to describing the system’s behavior.
‘These molecules have interesting electronic properties and are candidates for electronic and opto-electronic applications.’
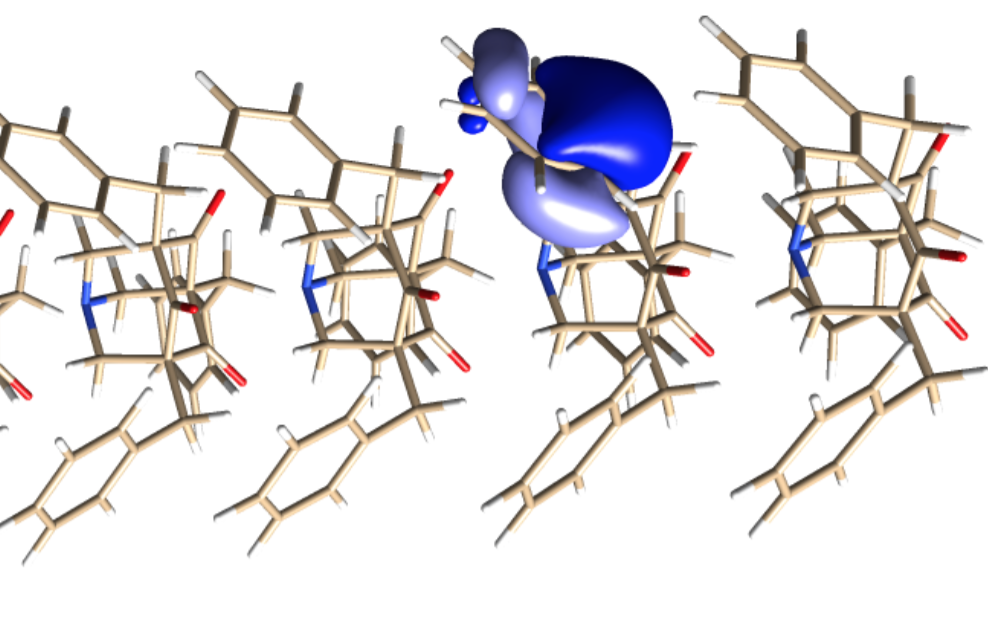
The supramolecular wire system thus provided a perfect test case for the experimental Divide-Expand-Consolidate (DEC) coupled-cluster (CC) numerical scheme, says Jacek Jakowski, an Oak Ridge computational chemist and one of the INCITE investigators. Since the calculations involve local interaction among electrons, they were amenable to division into lots of smaller calculations, as the DEC scheme was designed to do. And in contrast to the well-known density functional theory (DFT) algorithms typically used to model large molecular systems, the coupled-cluster (CC) numerical method incorporates these weak forces into its energy calculations.
The group simulated a supramolecular wire composed of 1-azaadamantane-trione (AAT) monomers. Azaadamantanes have asymmetrical ends that act as electron donors and receptors attracting each other into LEGO brick-like nesting configurations to form linear supramolecular wires.
“We are interested in understanding how these molecules assemble in solution and how the process of assembling can be controlled and directed,” Jakowski says. “These molecules have interesting electronic properties and are candidates for electronic and opto-electronic applications, such as organic light-emitting diodes, electric rectifiers and electro-optical modulators.”
The DEC strategy recently set a world record for this kind of simulation, modeling up to 40 monomers (2,440 atoms). The model kept 14,952 of Titan’s 18,688 nodes occupied for almost 19 hours, says a research report submitted to the prestigious Gordon Bell competition at the SC16 conference this fall in Salt Lake City. These calculations, which describe individual molecular orbitals, are well known but have never been modeled on such a large molecular system.
The key to achieving the computational breakthrough was dividing the molecular system into smaller fragments that could be calculated separately in the massively parallel computer and then recombined.
“For the parallelization, we used three hierarchical levels,” Jakowski says. “On the coarse level, the large molecular system is divided onto molecular fragments. This allows for independent-tasks parallelism. On the medium level, each of the independent tasks is computed in parallel using distributed memory.” At the fine level, the team used the OpenMP (open multiprocessing) programming interface and processor multi-threading to handle basic math operations.
The group’s initial INCITE award provided the resources to write the necessary code allowing the DEC algorithm to run efficiently on Titan and to benchmark it against some well-understood simulations for validation.
Thomas Kjærgaard, an Aarhus post-doctoral chemist and the report’s lead author, says describing electron-electron interaction depended on using local functions, files that divvy programs into smaller tasks. For the computationally savvy, “this may seem natural, but it is far from simple. The functions that naturally appear in the math are delocalized and span the entire molecule. Finding an efficient method to obtain these local functions for large molecules was the first of many breakthroughs required to achieve our goal.”
With their second INCITE allocation, now underway, they want to demonstrate that coupled cluster computational methods can be used for other complex molecular systems containing thousands of atoms. They hope to understand more fully the role of weak van der Waals forces on the structure and self-assembly of supramolecular wires.
Eventually, the research team expects the code will be used to benchmark other, less rigorous computational approaches such as DFT. In addition, they expect it will be combined with DFT to solve complicated surface catalysis and biochemistry problems. For instance, they suggest that an enzyme in solution might be modeled by calculating the active site and backbone with DEC while using a more traditional technique to model the solvent portion.
The researchers anticipate DEC will be useful for interpreting experimental results and, ultimately, to make reliable structural predictions. In the case of the nanowires, scientists would like to tune them to perform a variety of operations, Jakowski says. The computational chemistry group is collaborating with the Center for Nanophase Materials Sciences, where the project grew from prior user projects with Ronald Castelano from the University of Florida.