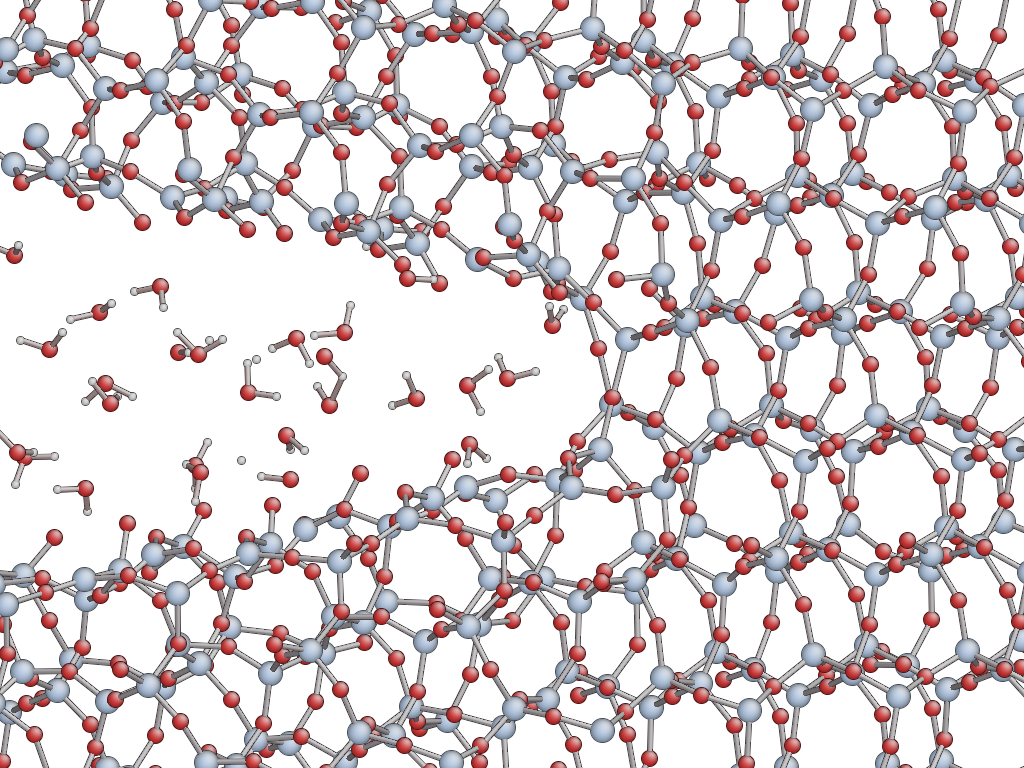
The chemical bonds that tie silicon to oxygen can be expensive to break.
Miners want to sever these bonds in great numbers. The bonds lock away valuable ores in rocks where the two elements are united in the form of silica. To extract small amounts of iron, copper, gold and other metals from large volumes of stone, the rocks must be ground up.
Grinding is costly. Mining in the United States uses more than 1,246 trillion Btu per year, a Department of Energy report says. That’s equal to all the electricity sold annually in Texas. The report also identifies the most energy-intensive part of the process and the one with the greatest potential for energy savings: grinding rocks.
“Breaking silica into bits uses huge amounts of energy,” says James Kermode, an assistant professor of engineering at the University of Warwick, Coventry, United Kingdom. “About 4 or 5 percent of human energy goes into smashing rocks up into tiny pieces. And in principle much less would be required if we understood everything that was going on.” He estimates that improving rock-grinding efficiency also could stop the release of millions of tons of heat-trapping carbon dioxide each year.
Unlike miners, most developers of new technologies want silicon dioxide bonds to stay intact. When silica breaks in medical devices, photovoltaic cells and buildings comprised of structural glass, the costs may be tallied not only in Btu or dollars but also in human health or lives.
“In some cases you want to prevent things from breaking,” Kermode says. “Dental implants, for instance, have to survive for really long periods of time in an aggressive environment, inside the human body, where there’s water and potentially other chemicals that could cause this stress-corrosion cracking – cracking that’s driven both by chemistry and by stress.”
To learn more about how these chemical bonds break, Kermode leads a study using Mira, an IBM Blue Gene/Q supercomputer at Argonne National Laboratory, to simulate the many ways silica crumbles. The long-term goal is to understand fractures thoroughly and then to develop, on one hand, tough new configurations for implants, electronics and construction and, on the other hand, mining techniques that are safer and more environmentally friendly than current methods.
Working with the Argonne Leadership Computing Facility, the team recently received a 2015 INCITE (Innovative and Novel Computational Impact on Theory and Experiment) award from the DOE Office of Science for 125 million processor hours on Mira to develop and run its models.
The award is a renewal of the team’s 2014 award, also for 125 million hours on Mira. The previous allocation produced a simulation showing how oxygen reduces the amount of physical stress needed to crack pure silicon. In the tip of a propagating crack, an oxygen molecule snaps apart. Its two liberated oxygen atoms work their way into the silicon crystal, applying heat that helps physical stress cleave a bond between silicon atoms.
‘You don’t compute the whole system quantum mechanically but only the part where some chemistry is happening.’
What’s more, the team directed its techniques to the rock-hard problems of silica, chemically called silicon dioxide. The researchers developed models of the atoms in this compound and used them to simulate the stress-induced fracture of silica in both its crystalline and glass forms.
Such models of these processes have become possible only recently, says team member Alessandro De Vita, a physics professor at King’s College London and Kermode’s former postdoctoral advisor. “It’s possible because we finally have good enough algorithms and we finally have good enough computers, like Mira,” he says.
The team had to meld sophisticated computational approaches to succeed. The most complete way to model a molecular structure would involve details of how the electrons and nuclei move. But this quantum mechanical (QM) modeling approach requires a huge number of calculations.
It’s been known for decades, Kermode adds, “that quantum mechanics provides very accurate models of many materials using a technology called density functional theory. The theory has been very successful in materials science. It’s just that the cost of these quantum mechanical calculations is very high. You can do a few hundred atoms or something like that, but you can’t do very, very large systems.”
So the team’s scheme uses quantum mechanics only at the crack tip, De Vita says.
“You don’t compute the whole system quantum mechanically but only the part where some chemistry is happening. That is a chemical bond which breaks and you need to represent it with quantum mechanics.”
The rest of the silica crystal, surrounding the crack tip, is a very, very large system. To model it, the team uses molecular mechanics (MM), which treats each atom as a ball and the bonds between them as springs. This approach uses the smallest number of calculations needed to exert the physical forces pressing or pulling on the overall crystal and creating stress at the crack tip.
This hybrid QM/MM multiscale simulation can capture details of stress-corrosion cracking – the simultaneous, combined effects of chemicals and physical force. This interplay is where the research team expects to find new insights into how chemical agents like oxygen or water conspire with stress to crack rocks and how future corrosive agents might reduce the energy needed to do the same.
This year, the team has added a machine-learning component as it continues modeling cracks in dry silica and in silica penetrated by water. The researchers managed to model a large system. Next, they had to make the simulation run over long time scales. Atomic-scale simulations require a calculation at every time step (a thousandth of a trillionth of a second), so a few trillionths of a second is a long time. Practically and philosophically, the researchers concluded they would not repeat such expensive calculations.
They saw a potential solution to the time-step problem in the repetitious configurations of an advancing fracture. “Nothing too much is changing as you go from one step to the next,” Kermode says. “History is repeating itself, and it all looks quite familiar. So we’d like to be able to reuse a lot of those expensive calculations. And that’s where the machine learning comes in.”
The INCITE team recently developed and published this part of the simulation scheme in the journal Physical Review Letters. The algorithm has assembled and is continually expanding a library of snapshots of molecules with breaking bonds. It compares each new step in the propagating crack with prior ones and, instead of redoing expensive calculations, uses any applicable previous ones. When a new configuration appears, the simulation does the necessary calculations, which also are saved in the library for later reuse. The team calls this component MLOTF (for “machine-learning on the fly”).
The group’s immediate goal is achieving large, multiscale simulations that can run over long time periods. “The technical focus is to develop new methodology for these kinds of systems – systems where there’s strong coupling between local chemistry and long-range stress,” Kermode says.